

Alzheimer, demans�n en yayg�n �e�idi olup d�nyada g�r�lme s�kl��� gittik�e artan bir n�rodejeneratif hastal�kt�r. En yayg�n erken semptomu k�sa d�nem haf�za kayb�d�r. Unutkanl���n yan� s�ra birtak�m bili�sel fonksiyon bozukluklar� da ortaya ��kar. Alzheimer’�n genetik ve �evresel fakt�rlerden etkilendi�i bilinmekle beraber hastal���n patofizyolojisi hala kesin olarak ��z�lememi�tir. Sinir h�crelerinin �l�m�ne neden olan iki temel hipotez vard�r: Ekstrasell�ler amyloid beta plak birikimi ve intrasell�ler n�rofibriler yumak. Son zamanlarda ara�t�rmac�lar Alzheimer’�n n�roenflamasyon ile ili�kisi �zerinde de durmaktad�r.
Amyloid Hipotezi
N�ronlarda transmembran proteini olan Amyloid Precursor Protein (APP); α, β, γ sekretaz adl� �� proteolitik enzim taraf�ndan belirli yerlerden kesilir. Alzheimer hastalar�nda APP ilk �nce, BACE1 ve BACE2 adl� en az iki farkl� kompleksten meydana gelen, β sekretaz enzimi ile kesilir. Daha sonra i�in i�ine γ sekretaz enzimi girer. γ sekretaz birden fazla yerden kesme i�lemi yapabilir. Bu i�lemler sonucunda ekstrasell�ler alana Aβ40 ve Aβ42 sal�n�r. Aβ42 daha uzun ve bir araya gelmeye daha yatk�n olan, suda ��z�nmeyen, n�rotoksik bir proteindir. Aβ42’ler bir araya gelerek amyloid plaklar� olu�turur. Alzheimer olmayanlarda α sekretaz enzimi, β-γ sekretaz� bask�lar. γ sekretaz�n katalitik fonksiyonlar�ndan sorumlu Presenilin1 (PSEN1) ve Presenilin2 (PSEN2) genleri vard�r. Bu genlerin mutasyonu Aβ birikimini artt�rmaktad�r.
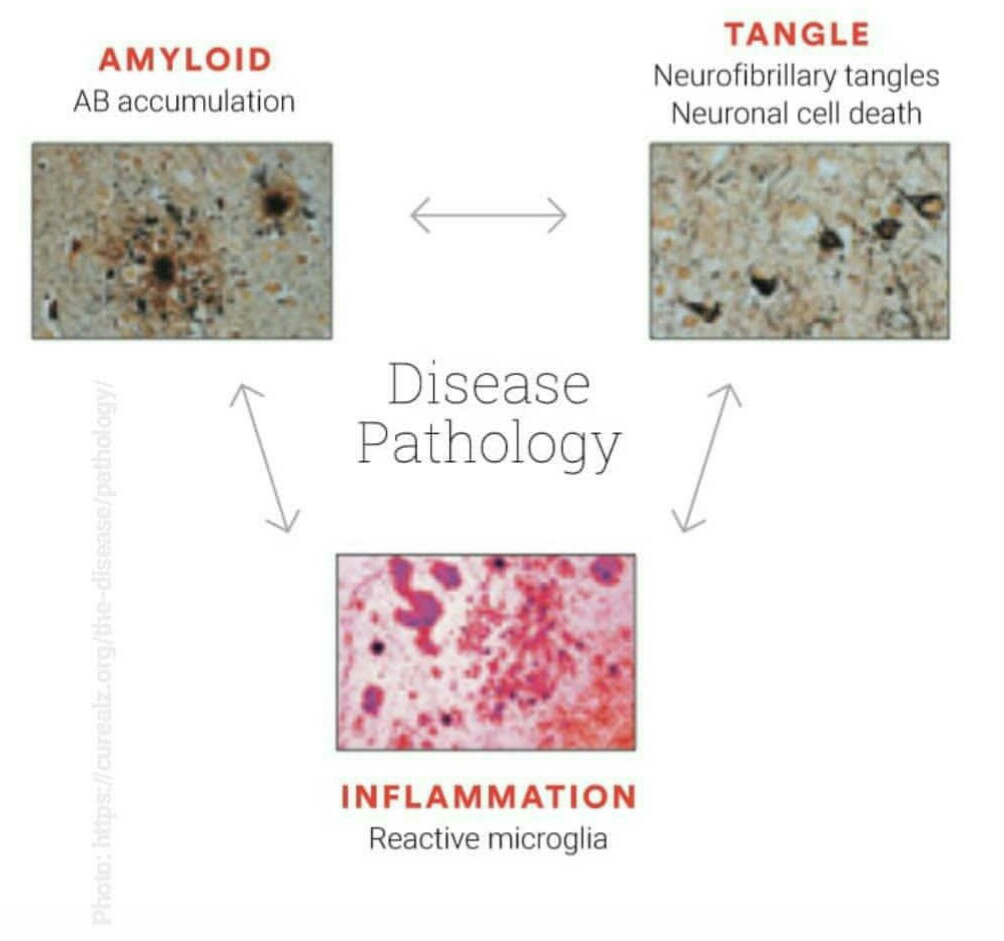
Tau Hipotezi
N�ronlardaki mikrot�b�lleri stabilize eden tau proteinleri, Alzheimer hastalar�nda hiperfosforile haldedir. Yeti�kin insan beyni her iki 3R ve 4R tau protein izoformunu eksprese eder. Alzheimer hastalar�n�n beyninde 3R ve 4R tau izoformlar� hiperfosforile halde iken birikir. Bu patolojik kal�nt�lar�n birikmesiyle n�rofibriler yumak olu�ur.
Alzheimer- N�roenflamasyon �li�kisi
Beyin imm�n h�creleri olan microglia’lar, beyindeki n�rolojik kal�nt�lar� (plaklar ve n�rofibriler yumaklar gibi) fagositik yolla temizler. CD33 geni, microglia’lar�n bu �zelli�ini etkin hale getirir. Ancak CD33 geni fazla eksprese edildi�inde microglia’lar�n sinir h�crelerini �ld�rd��� ve n�roenflamasyona neden oldu�u g�zlemlendi. Daha sonra CD33’�n tam tersi i�levi olan TREM2 adl� ba�ka bir gen ke�fedildi. TREM2, microglia’lar�n n�roenflamasyonu artt�rma etkisini engellemektedir. CD33 ve TREM2, enflamasyonu IL-1 beta ve resept�r� IL-1 RN’nin etkinli�ini artt�r�p ya da azaltarak d�zenlemektedir.
Alzheimer ve Diabetes Mellitus Tip 2 Aras�ndaki �li�ki
Tip 2 diyabet; ins�lin direnci, hiperins�linem ve bozulan ins�lin mekanizmas� ile karakterizedir. �ns�lin/�ns�lin benzeri b�y�me fakt�r� (IGF) –merkezi sinir sisteminde- sinaps olu�umu, n�roplastisite, ��renme, haf�za, n�ral k�k h�cre aktivasy�nu, akson uzamas� ve onar�m� gibi fonksiyonlarda �nemli rol oynamaktad�r. �ok say�da ara�t�rma ins�lin direnci ve eksikli�inin Alzheimer patolojisinde etkili oldu�unu g�sterdi. Alzheimer ve tip 2 diyabet; ins�lin duyarl�l���n�n bozulmas�, amyloid β birikimi, hiperfosforile tau proteinleri, vask�ler hasar ve n�roenflamasyon gibi bir�ok ortak patolojik �zelli�e sahipler. Hatta Alzheimer’�n Tip 3 Diyabet oldu�u g�r��� vard�r. 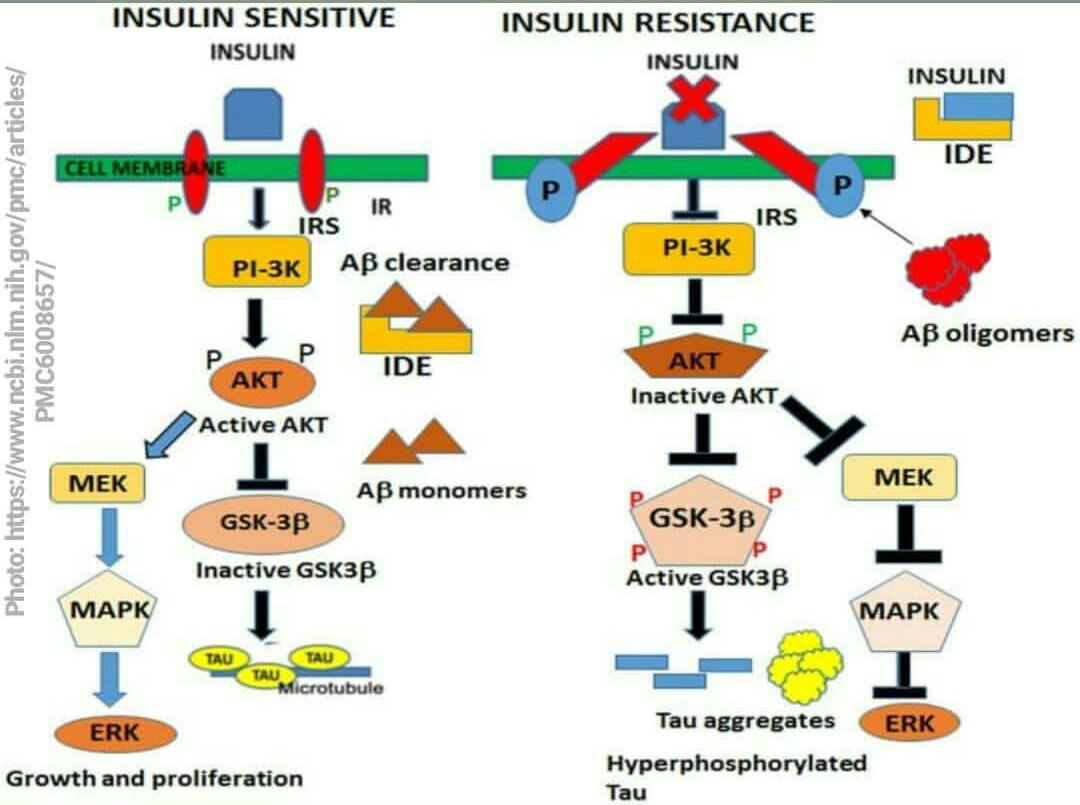
Baz� ara�t�rmac�lar Alzheimer beyninde, ins�lin resept�r� (IR), ins�lin benzeri b�y�me fakt�r�-1 resept�r� (IGF-1R) ve ins�lin resept�r substrat-1 (IRS-1) proteinlerinin eksprese edilmesinin ve aktivasyonunun azald���n� saptad�. Aβ oligomerleri, Alzheimer’da n�rodejenerasyonun ilerlemesinde katk�da bulunur. Hipokamp�steki n�ronlar�n dendritlerinde bulunan ins�lin resept�rlerini otofosforilasyon ile inhibe eder ve �nemli miktarda resept�r�n seviyesini ve aktivitesini azalt�r. Ayr�ca TNF-α/JNK yola��n�n etkinli�iyle IRS-1’i inhibe eder.
�ns�lin direnci, Aβ birikimini ve Aβ’y� olu�turan γ sekretaz gibi enzimlerin say�s�n� artt�r�r. �ns�lin ve Aβ, ins�lin degrade edici enzimin substrat�d�r, bu enzim her ikisini de par�alamakla g�revlidir. Hiperins�lineminin, ins�lin degrade edici enzimi bloke ederek Aβ y�k�m�n� azaltt��� �ne s�r�lm��t�r.
�ns�lin eksikli�i, n�rodejenerasyonu tau fosforilasyonu ile de artt�rabilir. �ns�lin ve IGF-1 tau fosforilasyonunu GSK-3β inhibisyonu ile engellemektedir. �ns�lin eksikli�i, Akt aktivitesini azalt�r ve bu da GSK-3β aktivitesini artt�r�r. Hiperfosforile tau proteinleri ve n�rofibriler yumak olu�ur.
Hiperins�lineminin, merkezi sinir sisteminde enflamatuvar yan�t� artt�rd��� bulundu. Periferik ins�lin seviyesinin artmas�, interl�kin1 beta (IL-1β), interl�kin-6 (IL-6) ve t�m�r nekroz fakt�r alfa (TNF- α) gibi proenflamatuvar sitokinlerin beyindeki seviyelerinin artt��� g�zlendi.
Bu zamana kadar olan �al��malar g�steriyor ki ins�lin direnci ve eksikli�i Alzheimer patolojisine olumsuz anlamda katk�da bulunuyor.
Yazar: Belemir UZUN, Ondokuz May�s �niversitesi T�p Fak�ltesi ��rencisidir.
KAYNAK�A:
Pathogenesis of Alzheimer’s disease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2685260/
Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications
Link between type 2 diabetes and Alzheimer’s disease: From epidemiology to mechanism and treatment
Cross-disease analysis of Alzheimer’s disease and type-2 Diabetes highlights the role of autophagy in the pathophysiology of two highly comorbid diseases
https://www.nature.com/articles/s41598-019-39828-5
Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer's Disease
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5797629/
‘Crosstalk’ between genes promotes brain inflammation in Alzheimer’s
https://neurosciencenews.com/alzheimers-genetics-inflammation-14461/
Alzheimer Hastal���n�n Geneti�i ve Epigeneti�i
https://dergipark.org.tr/download/article-file/292279
5 Comments
Ay�enur Y�lmaz
06 August 2019 12:38Talha
06 August 2019 12:55Mehmet Kaya
06 August 2019 14:20Bayram
07 August 2019 14:16Tuzlusuilegargara
08 August 2019 22:11