



Pulmonary Fibrosis
Pulmonary fibrosis is a hardening and scarring of lung parenchyma, caused by preceding inflammation either initially triggered by infection, environmental hazards, other systemic causes or idiopathic (unknown cause).
Know more about some of the conditions leading to pulmonary fibrosis
Cystic fibrosis is a multisystem and common life-shortening autosomal recessive disorder, affecting lungs (leading to pulmonary fibrosis), gastrointestinal tract, reproductive organs, salivary gland and others. In its pathophysiology, there is gene mutation in CFTR thus impedes chloride metabolism. Some of the presenting symptoms are recurrent pulmonary infection, productive cough, pancreatic insufficiency and elevated sweat chloride.
Idiopathic pulmonary fibrosis (also known as cryptogenic fibrosing alveolitis)
This is a form of chronic, progressive, fibrosing interstitial pneumonia of unknown cause but resembles usual interstitial pneumonia. In the past, its treatment aimed at minimizing progression from inflammation to fibrosis. However, underlying lesion of IPF may be fibrotic than inflammatory, explaining why few patients respond to anti-inflammatory drugs and prognosis remains darker
Pulmonary viral and bacterial infection
Pulmonary fibrosis can develop following inflammation caused by viral and bacterial infections for instance, in untreated or poorly treated pulmonary tuberculosis, inflammation and scarring left by mycobacterium tuberculosis often lead to pulmonary fibrosis before death.
Occupational and environmental causes: Organic and inorganic dusts, fumes and some gases
These dusts (e.g.: asbestos, silica.), fumes and gases when inhaled may lead to pulmonary fibrosis, for instance in mining, those tiny particles inhaled by miner often lead to sarcoidosis. Sarcoidosis in its nature encompasses fibrosing lung parenchyma, cough, shortness of breath and chest pain secondary to continued inflammation over time, caused by build-up of notorious tiny mineral particles in the lungs. Some gases and fumes over long exposure do the same thing.
Pulmonary fibrosis may be iatrogenic
Iatrogenic in this context means resulting from treatment/therapy complication. Though rate and frequency are not well defined; some anticonvulsants, chemotherapy agents and radiotherapy may induce pulmonary fibrosis as a long term adverse effect.
Pulmonary fibrosis may be genetic/inherited
In some conditions such as familial pulmonary fibrosis and Hermansky-pudlak syndrome, the lesion was found to be genetic or inherited to offspring.
Connective tissue/Autoimmune disease
Some forms of pulmonary fibrosis are sequelae of autoimmune conditions such as scleroderma, Systemic Lupus erythematosus, Progressive systemic sclerosis and others.
Signs and Symptoms
Symptoms of pulmonary fibrosis (PF) are gradual and non-specific from mild to debilitating form. Some of the symptoms at the onset may be fatigue and weakness, chronic dry and hacking cough. Very severe signs and symptoms are discomfort in the chest, clubbing of fingers, shortness of breath and oxygen desaturation requiring oxgenotharapy.
How can be a PF be diagnosed?
It requires an expert physician to diagnose the pulmonary fibrosis through history and physical assessments of the patient along with paramedical investigations. In a patient presenting the above- mentioned signs and symptoms, medical history should detail any occupational, environmental and familial predisposing factor that might have led to the development of the disease. Physical examination will be the whole patient with much emphasis on respiratory system.
In paramedical investigations, arterial blood gas (ABG) will be performed to measure blood pH, oxygen saturation and carbon dioxide content showing the ability of the lung to oxygenate blood and to eliminate carbon dioxide .Bronchoscopy will help to examine main airways in the lung using a small, flexible camera tool to visualize inside the lungs. Goal will be to evaluate problems in the lungs or blockage and to have easy access to sample such as tissue or fluids for further testing. A routine chest x-ray can be performed. However, X-ray may give some information but it cannot diagnose/confirm pulmonary fibrosis alone. High-Resolution Computerized Tomography (HRCT): It details image of the lungs to help physicians more clearly identify certain radiographic patterns in the lung tissue that may indicate disease.
A radiologist may identify a “honeycombing” pattern that suggests lung scarring and damage to the air sacs, or “ground-glass opacity”that refers to the hazy appearance of lung tissue that is most associated with inflammation. 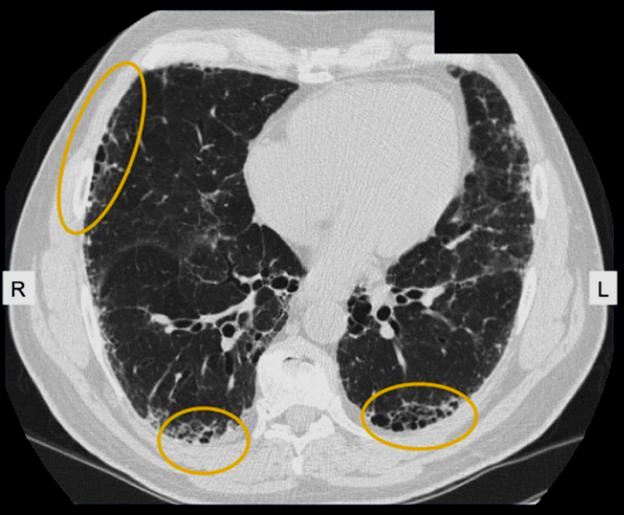
Pulmonary Function Tests: Breathing tests that measure the total amount of air in the lungs and assess the flow of air in and out of the lungs. There are two important components to pulmonary function tests: (1) spirometry that measures inspired and expired lung volumes and the rate at which this occurs and (2) diffusion capacity that measures the ability of oxygen to diffuse into the bloodstream. These tests are usually done in a hospital or clinical laboratory and consist of breathing into a spirometer; they are sometimes done sitting inside a large plastic enclosure that resembles a glass telephone booth. Some other tests such as bronchoalveolar lavage, surgical lung biopsy and exercise testing may be performed.
Treatment of Pulmonary fibrosis
Depends on whether the cause is known or not. If the underlying cause is infectious, meaning viral or bacterial: antiviral drugs and/or antibiotics can be among the treatment options If the cause is non-infectious other drugs such as prednisone, Cyclophosphamide, Azathioprine (immunomodulator), N-Acetylcysteine, Nintedanib: Nintedanib is an anti-fibrotic drug that is approved to treat IPF in the United States. Sometimes if it is too late, it requires lung transplant. Otherwise there is no success to cure disease rather managing symptoms will be the only option.
Prevention
Regular medical check-up, wearing mask and air filters for miners other protective occupational tools that prevent from inhaling dusts and other occupational hazardous substance.
Written by Dr Felix MUSABIREMA, MD
Currently Deputy Medical officer at UNITED NATIONS IN PORT-AU-PRINCE, HAITI
Former Medical Doctor at Ministry of Health Rwanda, Butaro Cancer center of excellence
References
1. www.connects.catalyst.harvard.edu/Profiles/display/Concept/Idiopathic Pulmonary Fibrosis
2. www.nejm.org/doi/full/10.1056/NEJMra1705751
3. www.lung.org/lung-health-and-diseases/lung-disease-lookup/pulmonary-fibrosis/
4. https://www.pulmonaryfibrosis.org/
0 Comment